Creating a new plugin: 8
Two issues
In the previous article, there were two issues mentioned and these are linked, so we'll fix them in this section. The first problem is that of the 'floating' oxygen atom at the end of the chain. This arises because when one amino acid links to the next, it loses a hydroxyl ion (-OH) from the carboxyl group in the molecule. If no link occurs, that ion is not lost and therefore there is an 'extra' oxygen atom to deal with. PDB files handle this by giving it a special name, 'OXT'. Of course, that atom is only present on one amino acid of the chain, and that could be any amino acid, depending on what type the last residue is.
There are two ways we could deal with this. The first is to add an extra bond to every amino acid in the bonds list file. That bond would be 'C' to 'OXT' and this would work fine; in all but the last residue the 'OXT' atom would never be found and so no bond would be made. The alternative would be to leave the bonds list unchanged but to look for an 'OXT' atom only in the last residue in the chain. Incidentally, we do need to check for it because not all PDB files seem to include the final 'OXT' atom, so we can't assume it's always there.
The first option would add an extra check to all the residues, which seems wasteful if we can find another way. The second method is the most efficient, but it requires that we know which residue is the last one in the chain. This leads on to the second issue. Since we've stored all the atoms in the entire protein molecule in one large array, we need to know which is the last residue for each chain to ensure that we don't create a bond between residues in different chains.
Finding the chains
Having a list of chains with the number of residues in each would solve both problems. We would know which was the last residue in each chain, so could avoid making bonds between that and the next one, and we could handle the terminal oxygen correctly. To do this, I created a very simple structure which contained the chain ID (a String) and the number of residues in that chain. Each chain structure is then added to another array called 'm_Chains'. But how do we know how many chains are in any molecule and where one chain starts and another ends?
Well, we could look at the PDB file. There is a record type called 'COMPND' which among other things lists the chain IDs. Take a look at this snippet of the first few lines from a PDB file:
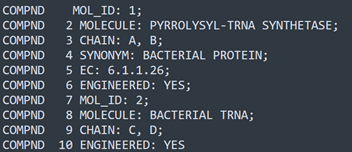
There are 10 lines of the COMPND record and one of these contains the word 'CHAIN' with a comma-separated list of chain IDs. But then there is a second line with CHAIN in it. This is because this file actually contains a protein molecule and a molecule of RNA (something else which we will have to handle!). There are four chains in total and we could get their IDs by parsing these few lines. But it's a fair bit of work and still doesn't give us the essential information: how many residues are there in each chain?
Or, we could look for 'SEQRES' records, which contain the residue sequence in each chain and the number of residues in the chain. The problem is that not all PDB files contain these records, so that's not helpful.
Each chain in the PDB file does contain a record named 'TER' at the end, indicating the end of a chain, so we could look for that. But it's not necessary because instead, once all the ATOM records have been loaded from the file, we know that each such record contains a chain ID value showing which chain it belongs to. So, if we iterate through the ATOM records array, all we need to do is get the chain ID from the first residue then count the number of residues until the chain ID changes. Then we start counting the residues in the second chain, and so on. This is very fast and at the end we know how many chains and how many residues in each chain without complex parsing of the CHAIN record lines or having to look for TER records.
Links between chains
Chains can be linked to one another with disulphide (sulphur to sulphur) bonds. There are only two amino acids which contains sulphur, cysteine and methionine (but disulphide bridges never involve methionine residues), and looking in a insulin molecule you can see that there are three areas where a disulphide link could occur (circled in green in the image below; bonds not shown, for clarity):
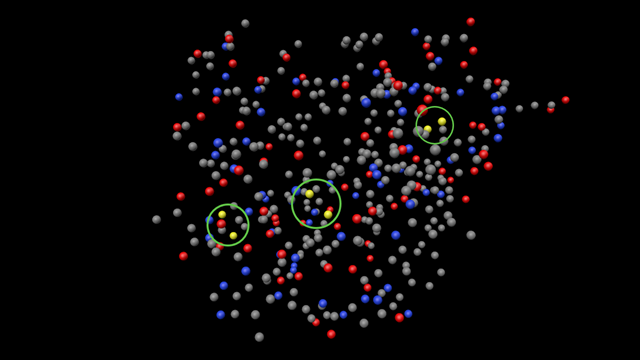
Fortunately the PDB file makes life a bit easier, because it can specify the sulphur-to-sulphur bonds in an 'SSBOND' record. There are three of these in the insulin PDB file, two between the two chains and another between two residues in chain A, which locks the chain into a specific configuration:
![]()
These lines only give the residues, so that the first one (for example) occurs between residues 6 and 11 in chain A. That's enough though, because all we need to do is find those residues, get the sulphur atom locations, and create a bond. This could be displayed differently in the scene display, e.g. by colouring the bond a bright yellow. Sometimes, these bonds are also shown in records named 'CONECT' but these records don't give the type of bond, so it could be between any atoms.
Fixing the OXT issue
With the chains listed we know the number of residues in each chain, so in the GenerateBonds() function if we count the number of residues as we process them we'll know when we come to the last one in a chain, like so:
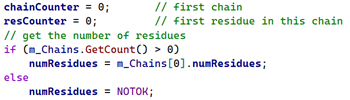
As the residue is processed, we store the position of the 'C' atom (to which 'OXT', if there is one, will always be attached) in a separate variable called 'termC' and for each residue the counter 'resCounter' is incremented. Then if this is last residue in the chain and we find the OXT atom, we store that in a second variable, 'termO', setting a flag called 'gotOXT' to indicate that we found the atom. All we do then is add this pair of points to the spline in exactly the same way as the peptide bond atoms:
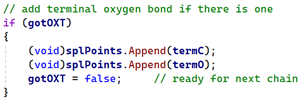
That solves the unconnected oxygen problem.
Handling multiple chains
Now we have an array of chains, this is really simple. When generating the bonds, we get the number of residues in the first chain, then count the residues as we process them. When we have processed the last one in that chain, we create a spline for that chain. Then we create another spline for the second chain in the same way, and repeat until all the chains have been processed. When complete, our GenerateBonds() function will return a null object with one or more child spline objects, each spline being one chain, and the null is returned to GetVirtualObjects. These are now separate splines and won't be linked together (except by the disulphide bonds, see above).
Processing multiple chains like this also lets us display different chains in different colours. For example, here is insulin showing the bonds only (no atoms) with the two chains in different colours:
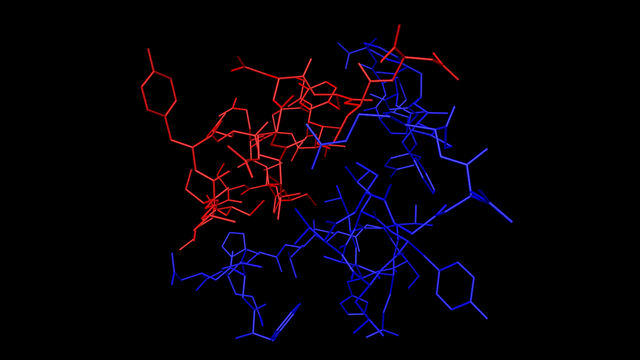
Or, here is an enzyme with 12 chains:
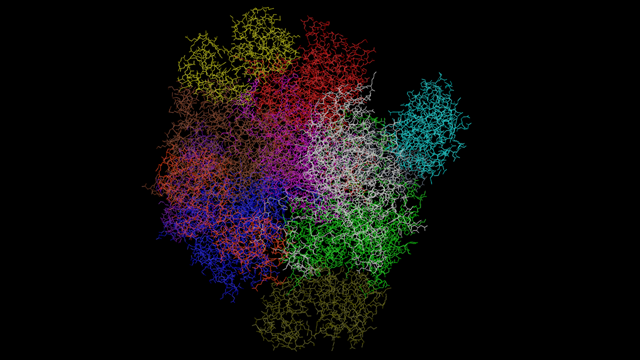
You can see how it's so much easier to visualise the topology of the molecule like this as opposed to looking at thousands of atoms. The only question to answer is how many different colours do we need? It turns out that most proteins have between 1 and 4 chains, but some have scores of chains and we can't provide an interface to handle all those. In any case, you have to wonder if a protein with, say, 60 chains could really distinguish between them with 60 different colours. So I've decided to limit the number of colours to 12, and if there are more chains than that, the 12th colour will be used for them. It's a compromise but it's also a trade off between maximum functionality and what can realistically be achieved in the interface.
Next time
There are still some issues to resolve. We need to be able to handle nucleotides as well as amino acids, since some proteins contain amino acid chains and nucleic acid chains. We still haven't added the disulphide bonds and I'm still considering whether to show single and double bonds differently. We'll look at in the next article.
Page last updated June 4th 2026