Creating a new plugin: 10
An extra feature (or two)
Before starting on nucelotides, there is an additional feature which I wanted to add. We can already colour all the bonds by a user-defined colour, or colour them by chain. What we can't do is colour them by nucleotide and this is what I will add now.
It turned out to be more complex than at first sight. The problem is that the method we use for generating the bonds for each chain results in a multi-segment spline dropped into a sweep object to create the geometry of the bond object. However, using only one sweep object might be efficient but it can only have one colour, making colouring the nucleotides differently impossible. The only solution to this is have a spline and a sweep object per nucleotide. I didn't want to do this because it increases the number of objects Cinema has to handle in the viewport, but there is no other way.
UI change
The first thing I did was add another option to the interface to enable colouring by residue, then in the GenerateBonds() function set a Bool named 'isResidues' to true if this option is selected. This means I can preserve the original method of generating bonds but add extra code to handle each residue separately.
As before, we iterate through all the atoms in the array, but instead of waiting until the end of a chain to create a spline, we create a spline at the end of each residue. Another required change is that when the new bond spline is created, we must pass the residue ID to the function which handles the spline creation. This is so the sweep object added for the residue can be given the correct colour, as determined by the residue ID.
And that raises a question: what colours to use? Traditionally these are seen in the RasMol protein visualisation app, and it seems pointless developing another set of colours. RasMol gives two possible schemes, the 'amino' and 'shapely' schemes, so we'll implement both of those.
Peptide bonds
The problem with the above method is how to include peptide bonds between residues. In the original code, these were all added in a block at the end of the array of points for the bonds within the residues, but we can't do that here. This was the trickiest part of the process. The 'N' atom in the first residue of the chain doesn't take part in a peptide link, and neither does the 'C' atom of the last residue. Worse, to make a link between residues we need the 'C' atom of one residue and the 'N' atom of the next residue. We could, in fact, colour peptide bonds differently to the residue bonds but that seems pointless, so I took the decision to colour the peptide bond the same as the bonds in the residue containing the 'C' atom of the peptide link. That is, if there are two residues in a chain, the first coloured red and the second blue, the peptide bond will take the colour of the first residue (red).
Getting the 'C' atom was done in the same way as the original method, except for adding a test to ignore that atom in the last residue in the chain. Getting the 'N' atom was more problematic. We already have a variable called 'atomCounter' which is an index into the atoms array and which always points to the first atom in a residue. As each residue is processed, this variable is updated to point to the first atom in the next residue, like so:
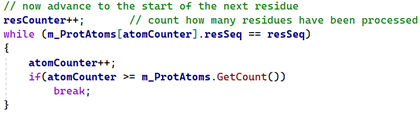
As long as the current residue is not the last one in the chain, we can use 'atomCounter' to get the first atom in the next residue, then iterate through that residue to find it's 'N' atom. In every PDB file I've looked at, 'N' is always the first atom in the residue, but that isn't guaranteed and given how...erratic these files can be, it's safer not to assume that will always be the case. So we have code like this:
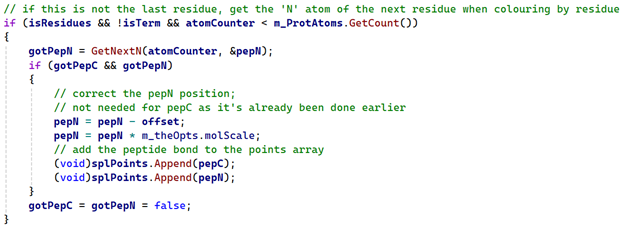
All the work here is done by the 'GetNextN' function, which looks like this:
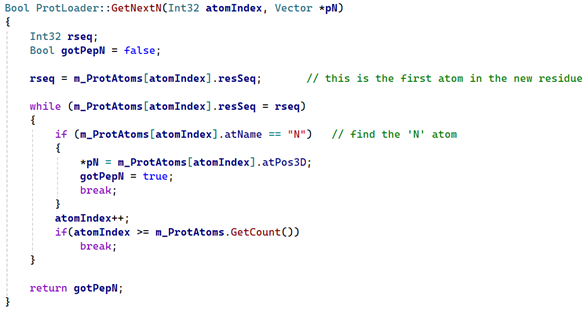
If the atom is found, the function returns 'true' and with the atom's position in the 'pepN' variable. If both the 'C' and 'N' atoms were found, the two vectors are then added to the points array.
Generating the spline
Finally, the function to create the spline itself and a sweep object is called, and the object returned inserted under the null object that GenerateBonds() will return. When creating the sweep object, all we do is pass the residue ID (a string such as 'GLY' or 'LEU') to a small function that returns the correct colour for that residue, which is then applied to the sweep.
The result
Here is glucagon again with each residue coloured using the 'amino' colour scheme:
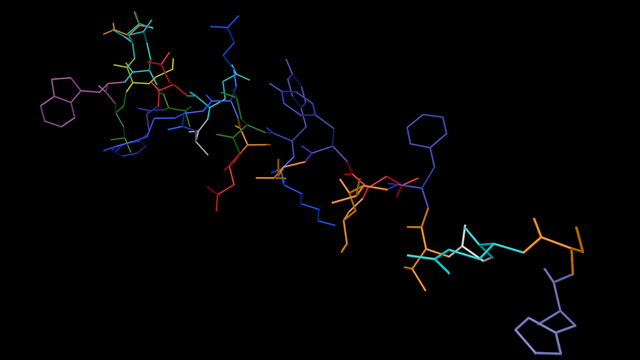
It would in fact be simple to convert the code so that the splines were always generated per residue, regardless of the colour mode, and I toyed with this idea because it would simplify the code used. However, a quick test shows that while there is subjectively no difference in the time taken to build the molecule and display it on screen, during animation there is a noticeable drop in the frame rate when colouring by residues. This is a consequence of generating many more objects in that mode compared to the single spline and sweep object method used originally. So, tempting though it is to simplify the code by only using the spline-per-residue approach, I decided to stick with the original method developed earlier except when colouring by residue.
We're almost done, but there is one other thing we can do. Take a look at this complex molecule where we colour the bonds by chains and display both bonds and atoms:

Although you can see that there are different colours for each chain, it isn't obvious because the atom colours obscure the bond colours. We could hide the atom display altogether so we only see bonds:

That's much better, but for the best results we can turn the atoms back on, but now add an override capability so that the atoms are coloured with their corresponding residue or chain colour like so:

I think this looks pretty nice. It won't always be wanted, but for maximum distinction between chains or residues, this gives the best results.
Page last updated June 12th 2026