Creating a new plugin: 2
Open a PDB file
So, we know what data we need from the PDB file - just the raw ATOM data to start with (see part 1 of this series). PDB files are simple text files, so all we need to do is open it and read each line (i.e. each record) from it. First though, there's a decision to make. Do we:
- load the entire file into memory so we can search through it to get the required data;
- or, read in one line at a time keeping only the data where the record type is ATOM.
There are pros and cons for each approach. The first method might be faster since we can read all the file at one go, rather than keep going back to it. But that will require more memory, though PDB files are typically only a a few hundred kilobytes long. It also means that if we need other data as well as the ATOM records, it's easy to find it from the stored file in memory without opening the file on disk and reading it again. On the other hand, reading one line at a time means that we can parse each record as it is loaded so that when the file loading is complete, we are ready to build the molecule.
I'm going to choose the second option, because as I'm just looking for atom data I don't need all the rest of the file. The test glucagon PDB file I'm using has 632 lines but 248 are data I don't want (yet, perhaps). But I won't parse each record on loading the file. I will keep all the atom records as strings in an array, because I will need to refer to them twice - for the atoms themselves and then the bonds between them.
However...
There's a snag. We could use the BaseFile class to open a file and read from it, using the BaseFile::ReadString() function. Unfortunately if you do this with an ordinary ASCII text file, two things happen. The SDK BaseFile functions assume that the file was created by Cinema, so the first four bytes are treated as a the file header, effectively discarding them. Then, the entire contents of the file are loaded into a single String variable! This isn't what is wanted at all. What we want is the equivalent of the C++ standard library getline() function, to read a line at a time - and there isn't one in the SDK. There are the InputStream functions, but again, there is no getline() function, just the ability to read the whole file or a specified number of bytes. Or, we could load the entire file into one String then iterate through it, looking for a newline character and separating it into lines when we find them.
For simplicity, I initially used the C++ standard library even though Maxon don't recommend it because it can throw exceptions. That worked but I felt uneasy about it and wanted to use the SDK only. If you look at the source of the getline() library function, all it does is read a file character-by-character until it hits a newline character. We can reproduce that. One thing we do have to watch for is the text line terminator. For files created in Windows, this will be a carriage return character and a line feed character ('\r\n'); for Unix and Mac OSX it's just a line feed ('\n') and for Macs pre-OSX it is a carriage return ('\r'). Since we don't know on what sort of machine the file was created, we have to check all these types when coming to the end of a line. Here is the working code:

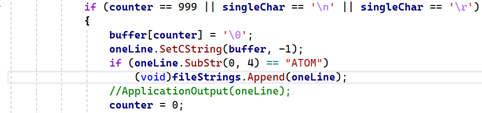
All this does is load one character at a time, storing it in a buffer, and stopping whenever we hit a line termination character or if the buffer capacity will be exceeded. The buffer needs to be null-terminated so that the String::SetCString() function will convert it into a String. It's just a matter of repeating this until we can't load any more characters. You might think this would be extremely slow, but thanks to the operating system and hardware disk buffers, it's actually very fast.
In the above code we simply print each line to the console. But it's a simple matter to store these strings in an array. If we create a class-level BaseArray and pass it to this function, which I did in the above code, we can store each line in its own String variable. It then becomes a simple matter to parse each line in turn to create the atoms.
The next thing is to keep only the lines we are interested in, which at the moment are the ATOM records. This code will do that; it's the same as before but checks that this is an ATOM record and keeps the whole string if it is:
Building the molecule
Now we have all the atoms, we will need to parse each line in the array, storing the data we need in a form we can use into an array of atoms. This will be the next part in this series. After that, we can easily build the molecule with the stored data.
Page last updated May 20th 2026