Creating a new plugin: 5
Displaying bonds between atoms
Now we have atoms, we need to display the bonds between them. There are different kinds of bonds we might want to show: single bonds, double bonds, peptide bonds (which are single bonds but we might want to display them differently) and disulphide bonds between peptide chains, if there are more than one.
None of which is difficult. All we need to do is create a spline to represent a bond, and this can then be rendered directly or dropped into a Sweep object to create geometry. But where are the splines located? We can draw them between atom positions, which we already know from creating the atom spheres. The problem is, which atoms connect to which? Unlike .mol or .sdf files, PDB files contain little or no details about bonds. In some files there may be a few CONECT records which specify a bond between two atoms, but these are usually few and are only present when there is a reason for them.
Take a look at this slice from the glucagon PDB file:
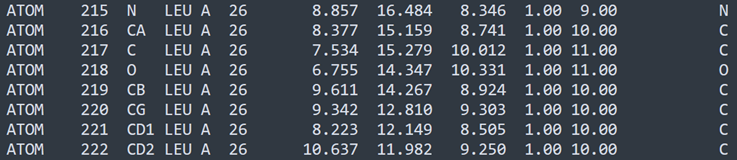
This is a single leucine residue. There are eight atoms (the hydroxyl ion which is part of the terminating carboxyl group is never included in the atoms list because it is lost when one amino acid is linked to another). We can see from the rightmost column that there are six carbon atoms , a nitrogen and an oxygen. You might guess that, in the order shown, the atoms simply bond to one another in a string, but that's definitely not the case. The leucine molecule looks like this (carbon is grey, oxygen red and nitrogen blue):
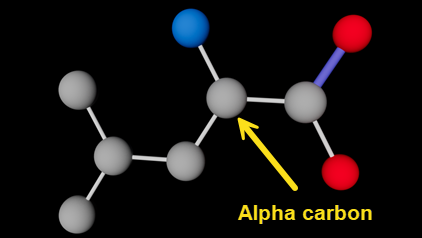
Leucine molecule (hydrogen atoms not shown); alpha carbon indicated
Which atom bonds to which other one(s) is the reason for giving each atom a name - these are the strings in the third column, so 'N', 'CA', 'CD1' and so on. Note that 'CA' is absolutely NOT the atomic symbol for calcium! To understand how this works, it's useful to know a couple of details about amino acid structure. All amino acids have an amino group (-NH2) and a carboxyl group (-COOH). Both these groups link to the same carbon atom known as the 'alpha carbon'. In fact, in the very simplest of all amino acids, glycine, that's all there is, as shown here with the alpha carbon indicated:
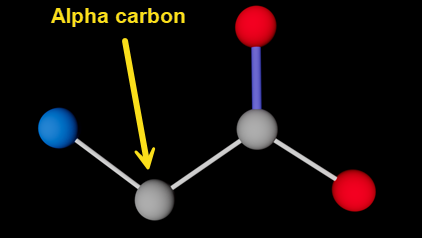
Glycine molecule (hydrogen atoms not shown); alpha carbon indicated
Atom names and their use
All amino acids other than glycine have a side chain, which also links to the alpha carbon. So, going back to the slice from the PDB file, it seems a fair guess that 'CA' is the alpha carbon - which is correct. 'N' is the nitrogen from the amino group, 'C' is the carbon of the carboxyl group, and 'O' is the oxygen in the carboxyl group, linked by a double bond to 'C'.
Fine. The other four carbons in a leucine molecule are the side chain. As you might expect, the first is 'CB', which links to 'CA'. But...then there is a 'CG' followed by two 'CD' atoms, 'CD1' and 'CD2'. Where do those names come from? Why aren't they in alphabetical order? In fact they are, but to understand this, there are two important principles: no atoms in the same residue can have the same name; and the first letter of the atom's name identifies the element (so 'CG1' would be a carbon, 'NZ' would be a nitrogen, and so on).
What happens when you have two or more atoms of the same type? This is where the trailing characters 'B' or 'G' or 'D1' come into play. The concept is that as an atom increases in remoteness - as measured by the number of intervening links - from the alpha carbon 'CA' it gets a suffix in alphabetic sequence. To make sense of this, you have to know that the sequence used is the Greek alphabet. This goes alpha, beta, gamma, delta...etc. These characters can' t be used in a plain ASCII file, so there are corresponding letters like so:
- alpha = A
- beta = B
- gamma = G
- delta = D
- epsilon = E
- zeta = Z
- eta = H
- ...and so on
This explains the letter sequence. The first side-chain carbon atom linked to 'CA' is 'CB'. Linked to that is 'CG' (not 'CC' as you might expect - Greek alphabet, remember). The next would be 'CD' but if you look at the image of the leucine molecule above, you can see that there are two carbon atoms linked to 'CG' and both have the same remoteness from 'CA'. Since they can't have the same name, they are named 'CD1' and 'CD2'.
I mention all this, not because a user of the loader needs to understand it, but because if you are trying to make sense of a PDB file it is so easy to become confused (as I did). For example, suppose an atom has the name 'NH2'. It is difficult at first to realise that this is NOT an amino group with one nitrogen and two hydrogen atoms, but a nitrogen atom at the 'eta' level of remoteness and that there is at least one other atom at the same level.
What do we do with all this information?
Once you understand all this, you can see how links can be made between atoms in the molecule. Fortunately, in the vast majority of cases the atoms in any given amino acid are the same in any protein, and the internal structure of the molecule is unchanged. How do we use that data to work out the bonds?
There are three ways it could be done. They are:
- Assume that atoms join to the nearest atom to them in physical space. We can work that out by checking the distance between atoms. With leucine, doing that would work fine, but for other molecules it might not, so bonds might not be made where they should or might be made where they don't exist in reality. Despite these problems, it may be necessary to do that if we have to handle molecules whose internal structure is not always known.
- Develop an algorithm that works out, depending on atom names, which other atom(s) they should link to. Again, this would work with leucine as it's such as simple molecule, but for others it might (would!) become extremely complicated.
- Since there are only 20 'standard' amino acids in proteins plus two others that are much less common, and since the internal structure of the amino acid is fixed, we could produce a lookup table of bonds in any amino acid. For example, for leucine that table would specify seven bonds - CA to N, CA to C, C to O, CA to CB, CB to CG, CG to CD1 and CG to CD2. All we would need to do for each residue is to get the list of bonds for that residue type, then for each bond get the positions of the relevant atoms in that specific residue in the PDB file, which would give us the two points of the spline representing the bond. The lookup table could also specify the type of bond - single, double, etc.
I'm going to use option 3, because although it involves the most initial work in determining all those bonds, coding the bonds is easiest that way. It does not, of course, take into account any other molecules in a protein that aren't amino acids, but we'll get to that later. So in the next part of this series, we'll code the addition of bonds in the amino acid residues themselves.
Page last updated May 26th 2026